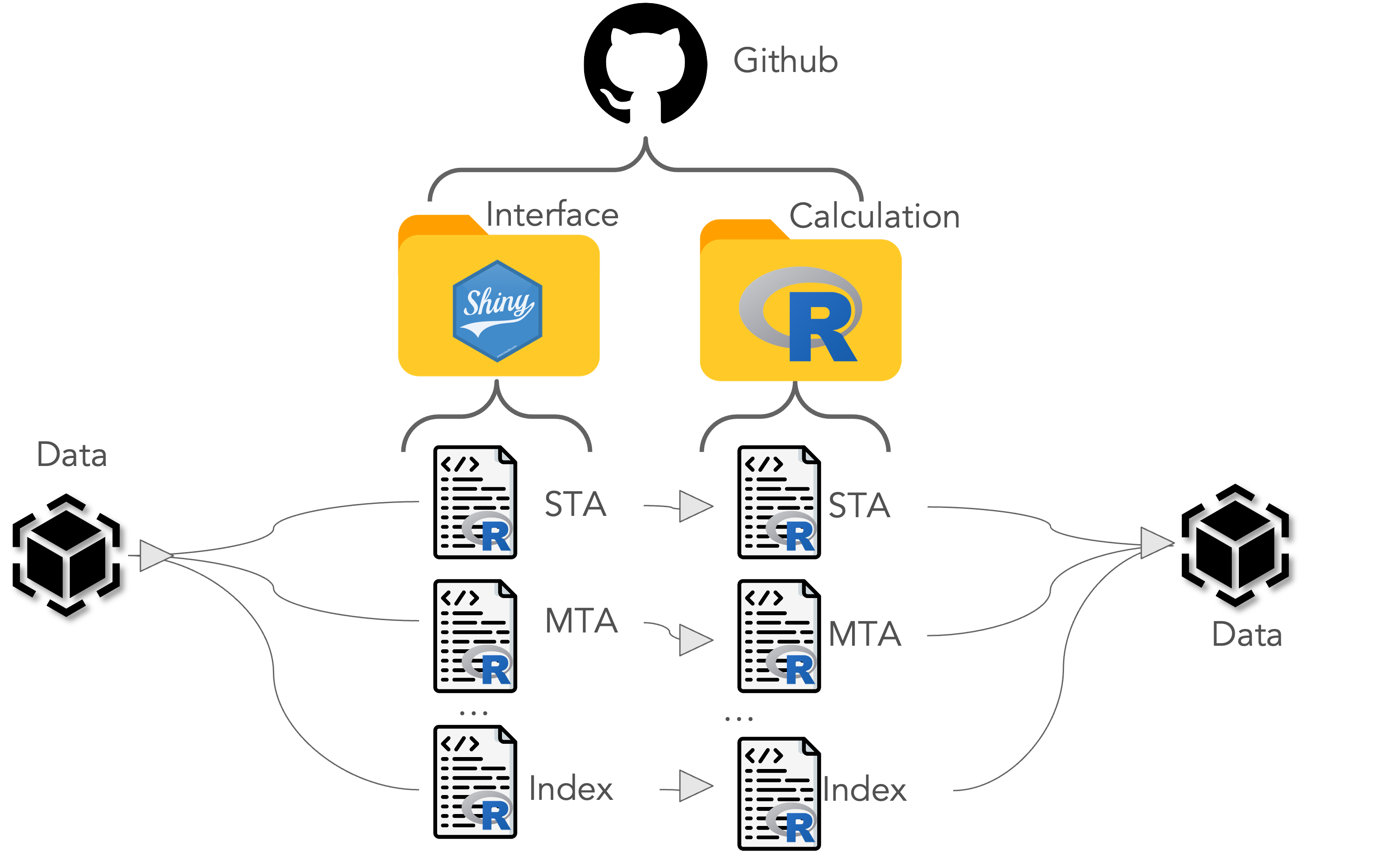
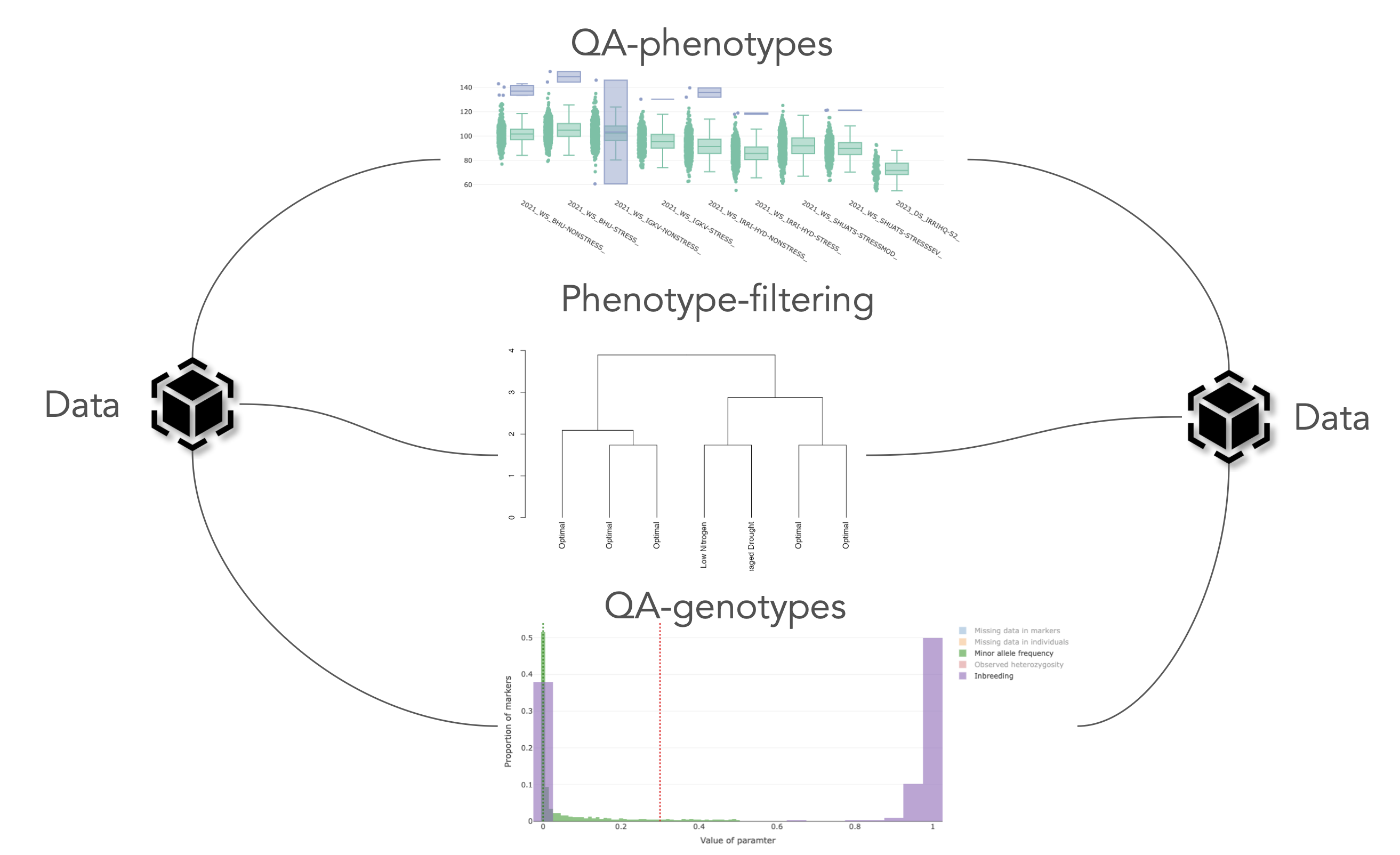
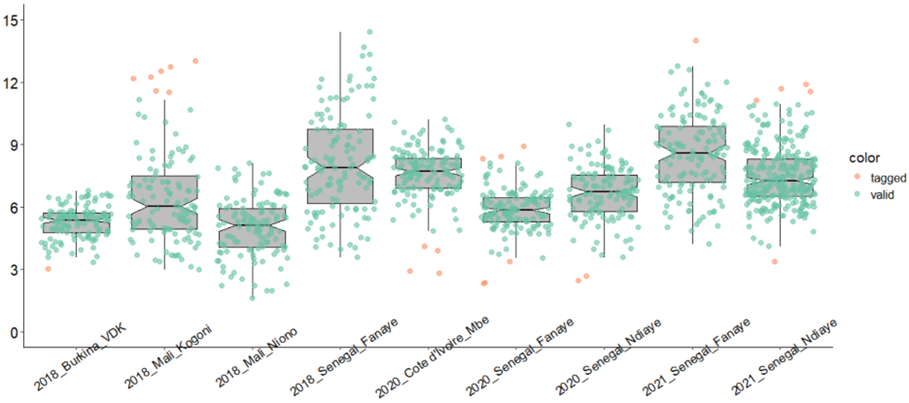
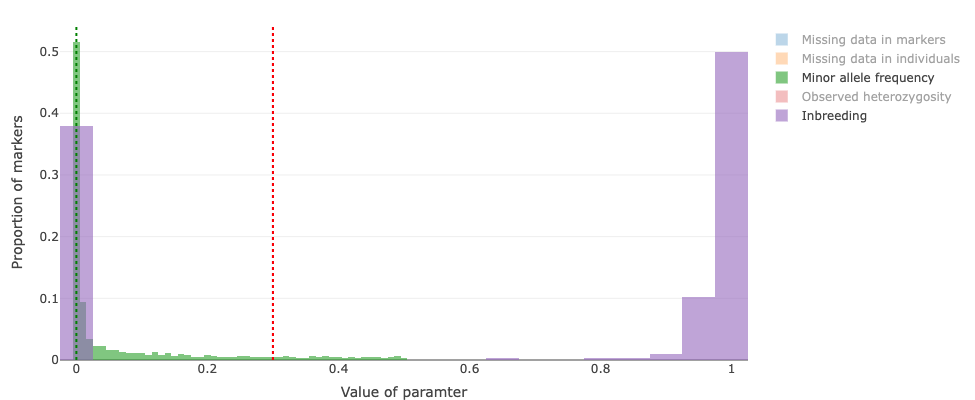
pipeline.-
The name of the column containing the labels describing the breeding effort to satisfy a market segment (e.g., Direct seeded late maturity irrigated).
stage.-
The name of the column containing the labels describing the stages of phenotypic evaluation (e.g., Stage 1, PYT, etc.).
year.-
The name of the column containing the labels listing the year when a trial was carried out (e.g., 2024).
season-
The name of the column containing the labels listing the season when a trial was carried out (e.g., dry-season, wet-season, etc.).
timepoint-
The name of the column containing the labels listing the timepoints from time series.
country.-
The name of the column containing the labels listing the countries where a trial was carried out (e.g., Nigeria, Mexico, etc.).
location-
The name of the column containing the labels listing the locations within a country when a trial was carried out (e.g., Obregon, Toluca, etc.).
trial.-
The name of the column containing the labels listing the trial of experiment randomized.
study.-
The name of the column containing the labels listing the unique occurrences of a trial nested in a year, country, location.
management-
The name of the column containing the labels listing the unique occurrences of a management (e.g., drought, irrigated, etc.) nested in a trial, nested in a year, country, location.
rep.-
The name of the column containing the labels of the replicates or big blocks within an study (year-season-country-location-trial concatenation).
iBlock.-
The name of the column containing the labels of the incomplete blocks within an study.
row.-
The name of the column containing the labels of the row coordinates for each record within an study.
column.-
The name of the column containing the labels of the column coordinates for each record within an study.
designation.-
The name of the column containing the labels of the individuals tested in the environments (e.g., Borlaug123, IRRI-154, Campeon, etc. ).
gid.-
The name of the column containing the labels with the unique numerical identifier used within the database management system.
entryType.-
The name of the column containing the labels of the genotype category (check, tester, entry, etc.).
trait.-
The name of the column(s) containing the numerical traits to be analyzed.